DMFT: solid_dmft and the effects of charge self-consistency
To study strongly correlated oxides with density-functional theory (DFT)+DMFT simulations, we rely on the triqs libraries. We put these powerful libraries together in the python wrapper solid_dmft [1, 2], developed by our group. We equipped solid_dmft with many functions, such as the calculation of DFT+DMFT energies, paramagnetic and (anti-) ferromagnetic systems and interfaces to Vasp and Quantum Espresso for charge-self-consistent DFT+DMFT calculations.
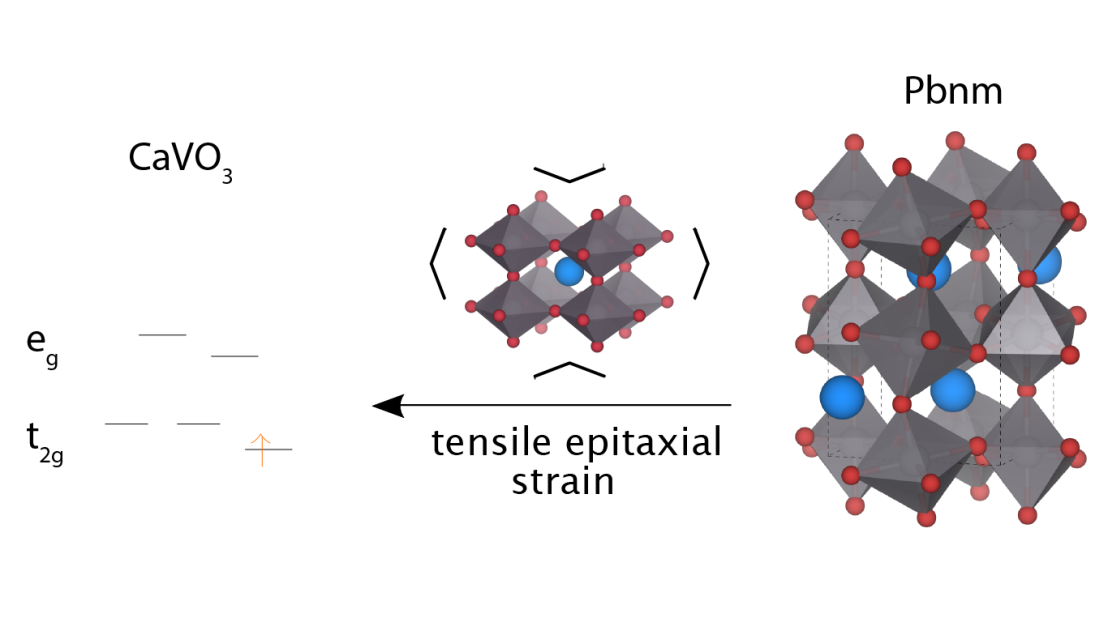
Using solid_dmft, we for example analyzed the effects of charge self-consistency on different systems such as strained CaVO3 [3]. We could show that technical details such as the so-called double-counting correction actually have a much more pronounced influence than the charge self-consistency itself.
However, if the system is close to phase transitions, the effects of charge self-consistency can be crucial to describe materials quantitatively. Additionally, charge self-consistency is mandatory for the total energy to be defined in DFT+DMFT, which is necessary for relaxing crystal structures.
- M. E. Merkel, A. Carta, S. Beck, and A. Hampel, external page solid_dmft: gray-boxing DFT+DMFT materials simulations with TRIQS, Journal of Open Source Software 7 (77), 4623 (2022)
- M. E. Merkel, A. Carta, S. Beck, and A. Hampel, external page GitHub repository of solid_dmft (2023)
- A. Hampel, S. Beck, and C. Ederer, external page Effect of charge self-consistency in DFT+DMFT calculations for complex transition metal oxides, Phys. Rev. Research 2, 033088 (2020)